
What’s in a (gene) name?
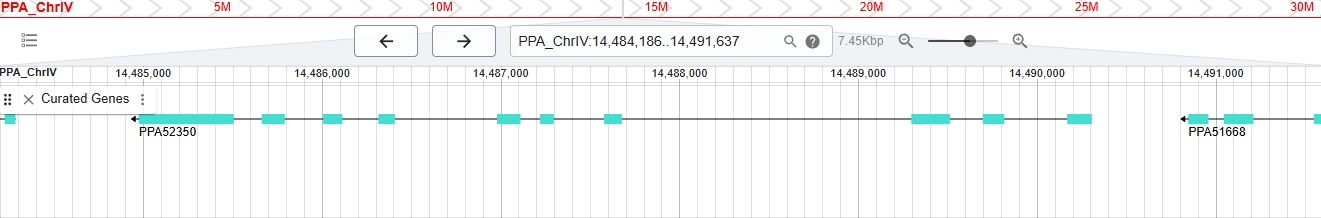
Screengrab from WormBase database showing exon and intron structure of a gene called PPA52350.
A chance conversation this week gave me a reason to check in on everybody’s favourite model organism database for nematodes…WormBase. Over two decades ago I spent four years of my life as a project manager for the UK arm of WormBase. Based at the Sanger Institute near Cambridge, we partnered with three groups in the USA (CalTech, CSHL and WashU) to maintain and develop the database that was used by thousands of nematode researchers around the world.
At the heart of WormBase was genetic and genomic data for the model organism Caenorhabditis elegans. This was the first animal to have it’s genome mapped and then sequenced. More impressively, the work to accurately fill in every last gap of the genome continued for many years after the formal publication of the genome sequence.
The 1998 Science publication described just over 19,000 protein-coding genes and a few hundred non-coding RNA genes.
I joined WormBase in 2001 and within a year or so I was tasked with developing what would frequently be referred to as simply ‘the new gene model’. Prior to any genome sequencing, there were many genes in C. elegans that had been defined by over two decades of classical genetic mapping approaches.
These genes were named in a simple, but consistent, way with three letters and then a number (this would necessarily have to be expanded to four letters in later years). E.g. unc-10 was the 10th gene named in the unc gene family (referring to UNCoordinated movements in worms with that mutation).
Enter the world of gene and genome sequencing
As the worm genome project began, genes would gain new identifiers based on in silico gene predictions made against the sequences of the cosmids, fosmids and YACs that would comprise the genome. So the first gene prediction on cosmid clone ‘T10A3’ would be named T10A3.1, the next adjacent gene prediction would be 'T10A3.2' and so on. Alternative splicing further complicated this nomenclature but it was relatively easy to append ‘a’, ‘b’, ‘c’ etc for splice variants. Luckily (for WormBase) I think 25 splice variants were the most ever discovered (see egl-8 in WormBase).
These sequence-based identifiers would sometimes become confusing when a new gene would be identified between the .1 and .2 gene predictions within a cosmid, fosmid or YAC. This messed up the original co-linearity of gene identifier with genomic location. E.g., you might end up with genes, F46H5.3 and F46H5.4 flanking a newly discovered gene which would gain a number such as F46H5.12 (you would use the next available number for that cosmid/fosmid/YAC).
When two genes go to war
The situation just continued to get more confusing. Turns out that a lot of computer gene predictions were not always right. This might mean removing a gene, or (more commonly) merging two genes into one new gene structure (where the original genes might now become splice variants. So genes C55B6.4 and C55B6.5 might become C55B6.4a and C55B6.4b (I’m using some made up examples here, but I recall it was somewhat arbitrary as to what gene identifier of the two merged genes would survive and which would die).
As genes were originally predicted on each assembled cosmid/fosmid/YAC sequence, and as these sequences overlapped — a necessary aspect of how the genome project was completed — it was possible that the same gene might be predicted independently on two overlapping cosmids/fosmids/YACs (and hence initially gain two unrelated gene identifiers).
Then of course there was the frequent situation where a classically defined gene would be matched with it’s sequence-identified counterpart. Hence, from my earlier example the unc-10 gene could also be known as T10A3.1.
Both names needed to be searchable and take you to the same entry in the database. But even more confusingly, there would be many more names that might exist. This could be because two researchers had independently mapped/published the same gene at different times, or just because some people would give the gene their own name without first referring to the literature. Generally speaking, the worm community is very good at avoiding this sort of thing but it did occasionally happen.
So unc-10 is not only the same gene as T10A3.1 but it could also be referred to as rim-1 or CELE_T10A3.1. I love that the WormBase database has preserved so many ‘curatorial remarks’ left by WormBase staff (myself included) as we tried grappling with how to merge genes and deal with other anomalies. E.g. here is a remark about the twk-18 gene:
”There are two twk-18 loci. One is CGC-approved (C24A3.6) and one is CGC-unapproved, which became an other-name of unc-58(T06H11.1a/b)”
This reflects yet another level of complexity where two different researchers or groups had used the same gene name for different genes.
Enter ‘the new gene model’
So it became my job to work out how we could:
- Create a new systematic identifier for every gene (yes, this reminds of me a bit of that XKCD comic)
- Roll this out across all of our parter groups in a way that didn’t break anything
This was a challenging problem but we eventually got there and all genes gained a new stable identifier that acted (hopefully) as a way to bring some order to how we (and others) worked with genes. These identifiers were ultimately rolled out in 2004 and an important part of ‘the new gene model’ was to allow a better way of capturing future gene births, deaths, mergers, and splits.
WormBase was based on ACeDB (A C. elegans DataBase), a bespoke database that was originally created by Richard Durbin and Jean Thierry-Mieg for the express purpose of managing C. elegans genetic data (it would go on to be used for many other organisms).
In AceDB you can always see the underlying model for any object by switching to something called the ‘Tree Display’. Amazingly (to me anyway), you can still do this in WormBase to the present day. It is buried within the Tree Display of genes, that you can see the original version control information that we added when we first migrated everything to ‘the new gene model’:

Screengrab from Tree Display view of a gene in WormBase showing the Version change information for a gene.
In this case ‘geneace’ was the local ACeDB database that we used at the Sanger Institute to store information regarding all of the classically mapped genes. I was (and perhaps still am?) ‘WBPerson1971'; this means I am forever indirectly associated with most of the initial gene set of C. elegans in WormBase! This now brings me to the fundamental point that I wanted to make with this very long blog post…
What was the format of the new gene identifier?
I remember wanting to borrow from the format that the Ensembl genome database had established. Gene identifiers should have a fixed width by using leading zeroes to pad out the identifier (this makes it much friendly to computer programs that have to process such data). I also wanted there to be a simple text prefix to the identifier (Ensembl has gene IDs such as ENSG00000139618: ENSG = ENSembl Gene).
I went with ‘WBGene’ for the prefix part which just left the question of how many digits to reserve for the gene space. At the time I was working on this I think the genome of a related nematode C. briggsae had been finished. As I recall, WormBase contained that data as well as some very limited data for a few other nematodes (including some classically mapped genes as well as many sequence derived genes). So I knew that WormBase would grow beyond the 20,000 or so genes there were at the time of the C. elegans genome publication.
I opted for eight digits in the identifier, allowing for 99,999,999 genes. At the time this did seem like overkill, but I would rather err on the side of caution than give future bioinformaticans major headaches, e.g. what if we had gone for a five-digit number and then it turns out that we needed to store over 100,000 genes?
All of this meant that our unc-10 gene from before could now also be known as WBGene00006750.
An end to WormBase and an end to ‘the new gene model’
This week I wanted to have a look to see what the highest gene identifier was that had been added to WormBase. In taking a look at the website, I was saddened to see that WormBase came to an end in July 2025 with the 298th release of the database.
Thankfully, most of the data will live on in the Alliance of Genome Resources which is a consortium of seven model organism databases. It’s not clear to me whether the alliance will end up with yet another tier of identifier that will span all of the species that are represented. I note that unc-10 in their database currently gains a slight tweak with an extra prefix of ‘WB:’ to become WB:WBGene00006750 (presumably because they have the extra challenge of needing to know which database an identifier came from).
I wonder whether the new gene model will live on in the Alliance of Genome Resources and whether those eight digits of reserved identifier space will continue to fill up. Given how easy (and relatively cheap) it has become to sequence genomes these days, someone might decide they want to sequence the genomes of all nematodes (there are about 25,000 described species, but there could be many more).
The last gene in WormBase
And so I can finally bring this post to a close with the reveal of the last gene identifier that made it into the last release of WormBase:
WBGene00311061 (also known as PPA52350) is a protein-coding gene from the nematode Pristionchus pacificus. It is the highest number gene that I could find and it reflects the fact that the gene count of WormBase has increased over 15-fold since my time there.
I bet this is due to a lot of other species being added, but also due to an explosion in RNA genes in C. elegans. I’m glad that my choice of gene identifier all those years ago has survived.
In writing this blog post, it’s been fun a lot of fun to take this trip down memory lane. In my research for this post, I came across a recent video (March 2026) by the Alliance of Genome Resources which explains much more about how worm and fly genes are named. Tim Schedl is the gene name curator for the WormBase data within the Alliance dataset who took over from Jonathan Hodgkin who I worked very closely with during my time at WormBase.
If there is are any lessons to be learned from all of this (especially to any young bioinformaticians out there), I would say:
- It’s hard to design databases for biological data. Biology is messy and will produce surprises that might only emerge years after you define a schema that thought would capture all possible edge cases (biology will then laugh at your schema)
- If you can, try to future proof things as much as possible
- Do not - under any circumstances - allow asterisks or question marks to be part of a valid gene name! There were originally some genes in the geneace database that included asterisks which caused all manner of problems in ACeDB as asterisks were also used as a wildcard search operator.




