
Tales of drafty genomes: part 3 – all genomes are complete…except for those that aren't
This is the third post in an infrequent series that looks at the world of unfinished genomes.
One of the many, many resources at the NCBI is their Genome database. Here's how they describe themselves:
The Genome database contains sequence and map data from the whole genomes of over 1000 species or strains. The genomes represent both completely sequenced genomes and those with sequencing in-progress. All three main domains of life (bacteria, archaea, and eukaryota) are represented, as well as many viruses, phages, viroids, plasmids, and organelles.
This text could probably be updated because the size of the database is now wrong by an order of magnitude…there are currently 11,322 genomes represented in this database. But how many of them are 'completely sequenced' and how many are at the 'sequencing in-progress' stage?
Luckily, the NCBI classifies all genomes into one of four 'levels':
- Complete
- Chromosome
- Scaffold
- Contig
I couldn't find any definitions for these categories within the NCBI Genome database, but elsewhere on the NCBI website I found the following definitions for the latter three categories:
Chromosome - there is sequence for one or more chromosomes. This could be a completely sequenced chromosome (gapless) or a chromosome containing scaffolds with unlinked gaps between them.
Scaffold - some sequence contigs have been connected across gaps to create scaffolds, but the scaffolds are all unplaced or unlocalized.
Contig - nothing is assembled beyond the level of sequence contigs
So considering just the 2,032 Eukaryotic species in the NCBI Genome Database, we can ask…how many of them are complete?
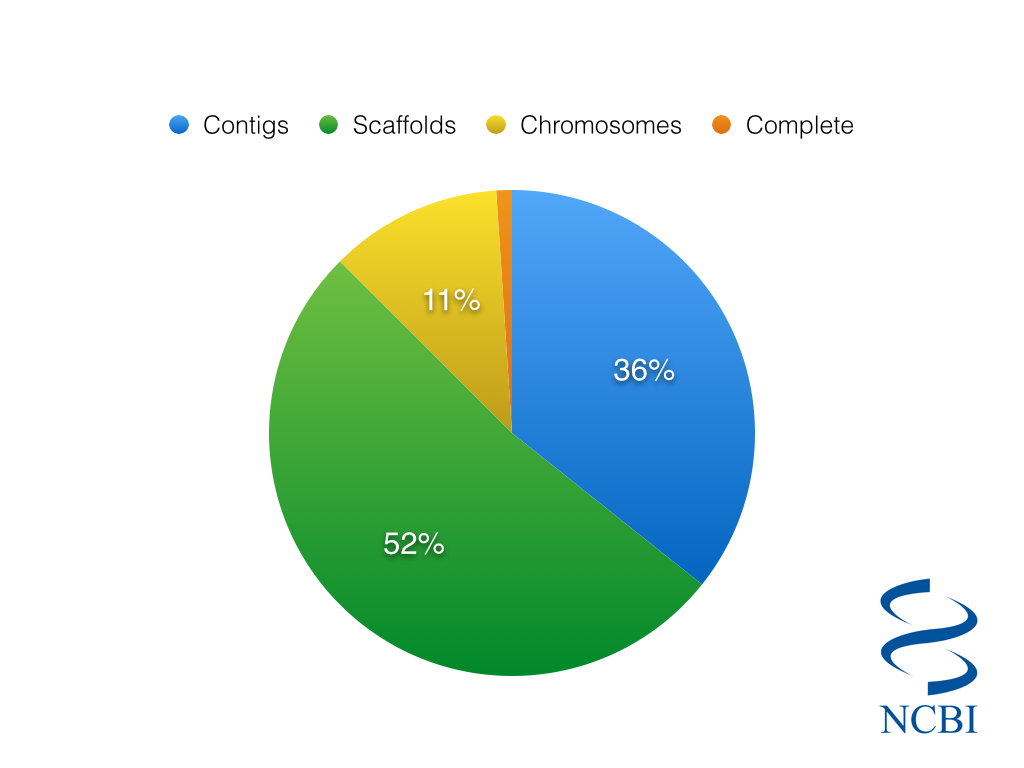
Completion status of 2,032 eukaryotic genomes, as classified by NCBI
The somewhat depressing answer is that only a meagre 24 eukaryotic genomes are listed as complete, about 1% of the total. Even if we include genomes with chromosome sequences, we are still only talking about 13% of all genomes. You might imagine that the state of completion would be markedly better when looking at prokaryotes. However, only 11.5% of the 31,696 prokaryotic genomes are classified as complete.
In the last post in this series, I included a dictionary definition of the word 'draft'. This time, let's look to see how Merriam-Webster defines 'complete':
having all necessary parts : not lacking anything
not limited in any way
not requiring more work : entirely done or completed
By this definition, I think we could all agree that very few genomes are actually complete.



